L’origine du SARS-CoV-2 : Partie 1
Emergence d’une nouvelle espèce

Image avec l'aimable autorisation de MammalWatching.com
Naturellement, l'une des premières questions que nous nous posons lorsqu'une nouvelle maladie apparaît est "d'où vient-elle ?" Dans cet article, nous parlerons des origines du SARS-CoV-2 et de la façon dont il a probablement émergé dans la population humaine.
Chaque nouvel agent pathogène émergent est accompagné d'une théorie du complot sur son origine. Le SARS-CoV-2 n'est pas différent. Dans la première partie de cette courte série, nous parlerons des données sur l'émergence de ce virus dans la population humaine. Dans la deuxième partie, nous approfondirons un peu plus et discuterons des raisons pour lesquelles ce virus est si efficace pour infecter les humains. Et enfin, dans la troisième partie, nous aurons tous les éléments nécessaires pour discuter et démystifier les nombreuses théories conspirationnistes qui entourent le SARS-CoV-2.
La première question à laquelle nous devons tenter de répondre est la suivante : quelle est l'origine la plus probable du SARS-CoV-2 ? Pour répondre correctement à cette question, nous aurons besoin d'un peu de contexte.
Animaux et virus mutants

Graphique décrivant la transmission des maladies zoonotiques et la manière dont différents vecteurs peuvent propager la maladie (avec l'aimable autorisation de l'OMS).
On estime que 75 pourcent des agents pathogènes humains émergents sont d'origine zoonotique. Le terme "zoonotique" désigne les maladies qui se transmettent des animaux aux humains (voir l'image ci-dessus). Chaque animal possède ses propres micro-organismes - virus, bactéries, etc. - qui sont devenus capables d'infecter cet animal en particulier. Par exemple, le virus de l'immunodéficience simienne (SIVcpz) infecte fortement les chimpanzés. Cependant, il est très mauvais pour infecter les souris. Le SIVcpz a du mal à infecter les humains, mais les scientifiques pensent que le SIVcpz a été le précurseur du virus de l'immunodéficience humaine (VIH-1). Au gré des passages à d’autres espèces, SIVcpz a commencé à acquérir des mutations qui lui ont permis de bien se développer chez l'homme, jusqu'à devenir une espèce distincte de virus—le VIH. Ce processus de transmission zoonotique et de mutation est important pour comprendre les origines du SARS-CoV-2.
Les micro-organismes sont conçus pour les animaux qu'ils infectent. Cependant, il est important de se rappeler que, dans la nature, ces micro-organismes ne se conçoivent pas de manière anticipée. Au contraire, ils s'agrippent au hasard, faisant des billions de copies d'eux-mêmes avec de légères différences à chaque fois. La majorité de ces mutations n'aident pas du tout et certaines peuvent même avoir des effets néfastes, mais ils finissent par tomber sur quelque chose de vraiment génial, et cela leur permet d'être plus efficaces. Ce processus s'appelle la sélection naturelle. Les virus se reproduisent (c'est-à-dire qu'ils se multiplient) de manière rapide et négligée, en faisant des erreurs mais aussi en améliorant leurs chances d'acquérir une mutation avantageuse qui surpasse toutes les autres.
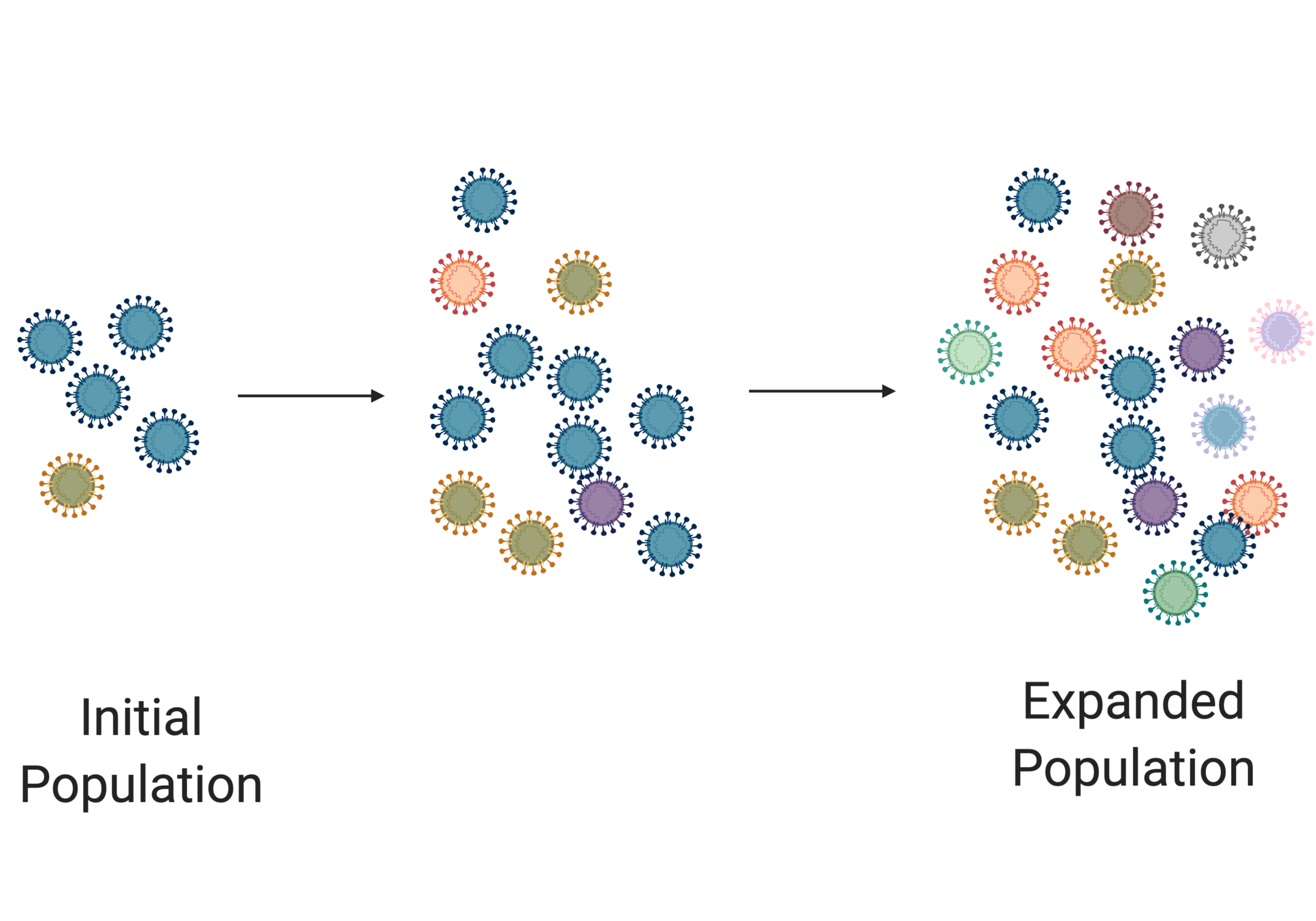
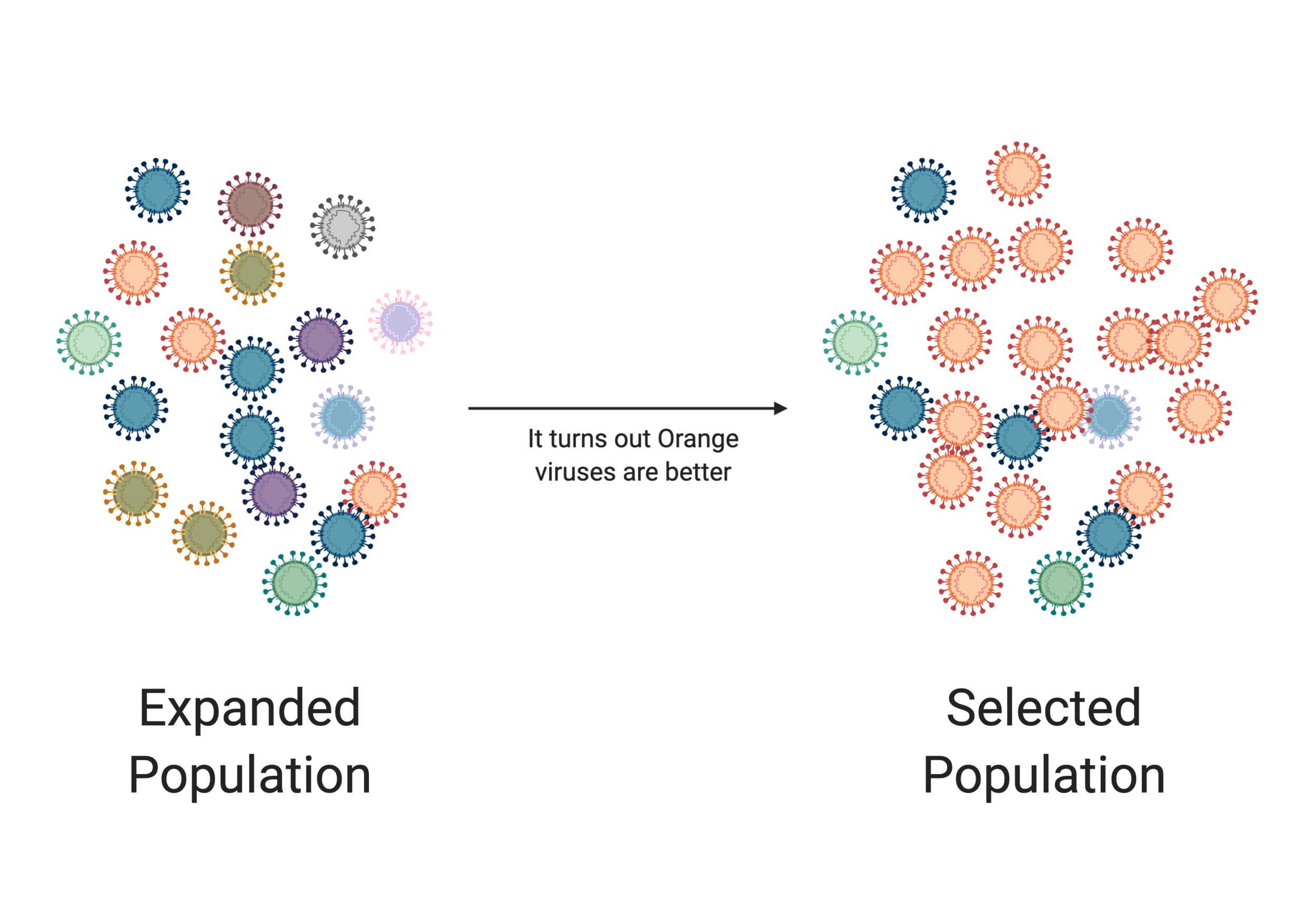
Sur l'image ci-dessus, nous voyons une population initiale de virus. Ils sont peu nombreux et peu diversifiés (ce qui est indiqué par leurs différentes couleurs). Mais à mesure que ces virus se reproduisent, ils commencent à faire des erreurs. Au fur et à mesure que la population s'accroît, nous constatons une augmentation de la diversité de la population de virus. Mais dans l'image suivante, il se produit quelque chose qui donne un avantage au virus orange (par exemple, il a sauté dans une nouvelle espèce). Dans ce scénario, le virus orange prend le dessus sur la population. Ce processus d'expansion et de sélection est vital pour l'évolution du virus.
La plupart d'entre nous ont déjà entendu parler de la sélection naturelle, mais il est important d'insister sur ce point. C'est à ce processus de mutation aléatoire, qui aboutit à des virus de plus en plus adaptés, que nous pensons lorsque nous essayons de déterminer l'origine d'un virus. Nous pouvons évaluer les informations qui sont contenues dans son génome à ARN, et chercher des indices sur son origine. Partout où un virus a été présent, sa signature est inscrite dans son histoire génétique.
Les premiers indices
Avant que les premiers cas de SARS-CoV-2 ne soient reconnus, il y avait 4 espèces de virus connues pour infecter les humains dans le genre Betacoronavirus (le même que le SARS-CoV-2 allait rejoindre). Les deux premiers, HKU1 et OC43, provoquent un simple rhume. Les deux autres, le SARS-CoV et le MERS-CoV, sont des virus extrêmement mortels qui ont provoqué de graves épidémies (mais pas de pandémie mondiale).


A gauche - les gros titres de 2003, lors de la première épidémie de SRAS. A droite - les gros titres de 2012, lorsque le MERS a fait son apparition. Les transmissions du MERS des chameaux et camélidés vers l'homme se poursuivent encore aujourd'hui.
Le 31 décembre 2019, le gouvernement chinois a signalé plusieurs cas de pneumonie virale dans la ville de Wuhan, en Chine. Beaucoup de ces cas étaient liés au marché de gros de fruits de mer de Huanan. La première crainte était que le SARS-CoV original soit de retour, ou peut-être qu'une nouvelle souche mortelle de grippe circulait. En une semaine, les scientifiques chinois avaient déjà soumis au monde entier la séquence génomique complète du virus. Nous avons appris qu'il ne s'agissait pas du SARS-CoV, mais d'un nouveau coronavirus initialement nommé nCoV-2019.
A ce moment, le monde savait 3 choses sur le virus :
Il pouvait provoquer une pneumonie virale potentiellement mortelle
Il était étroitement lié au SARS-CoV
De nombreux cas, mais pas tous, étaient liés au marché de gros de fruits de mer de Huanan

Il s'agit d'une civette, l'hôte intermédiaire présumé entre les chauves-souris et les humains pour l'épidémie initiale de SRAS de 2003. Avec l'aimable autorisation de Kalyan Varma et de Wikimedia Commons.
Les recherches menées lors de l'épidémie initiale de SARS-CoV ont montré que le virus s'était probablement répandu chez l'homme à partir de Rhinolophes via un hôte intermédiaire, la civette masquée (photo de droite), avant de sauter sur l'homme. Les scientifiques ont donc émis l'hypothèse que le nCoV-2019 (aussi appelé SARS-CoV-2) s'est également répandu chez l'homme à partir des chauves-souris, peut-être en utilisant un hôte intermédiaire différent.
Bien que cette hypothèse soit prometteuse, nous avons encore besoin d'un moyen de la tester. Cela nous amène à la prochaine question importante : comment déterminer les origines d'un virus ?
Les outils d’un détective de virus
Identifier les origines d'un virus est un jeu passionnant de déduction holmésienne, de puzzles logiques et d'inférences. En tant que détectives de virus, nous commençons par rassembler autant de données et de preuves que possible. Malheureusement, il n'y a souvent pas de preuve irréfutable, et nous devons donc utiliser le raisonnement inductif pour arriver à des conclusions sur des événements que nous n'avons pas observés directement.
Ainsi, si nous ne pouvons pas observer un virus au moment où il est transmis à un humain pour la première fois, comment pouvons-nous voir d'où vient un virus ? Nous le soumettons au vieux traitement 23andMe et nous examinons son génome pour comprendre ses ancêtres.
N'oubliez pas que au fur-et-à mesure que ce virus fait des copies de lui-même, il peut faire des erreurs. Certaines de ces erreurs vont nuire au virus et les virus contenant ces erreurs vont échouer. Certaines de ces erreurs aident le virus et ces virus se développeront abondamment. Certaines de ces erreurs n'auront aucun effet sur le virus. Nous appelons ces mutations neutres et, par conséquent, tous les virus qui en sont les enfants contiennent désormais une mutation de signature indiquant leur parenté. Nous pouvons utiliser ces signatures pour construire un arbre généalogique.
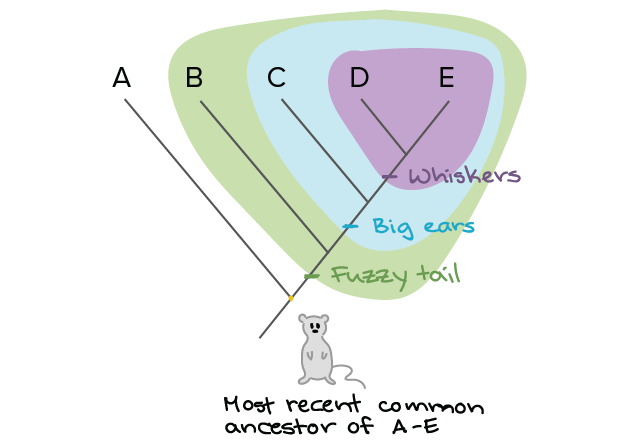
Une illustration d'arbre phylogénétique, avec l'aimable autorisation de l'Académie Khan
Comme illustré ci-dessus, nous pouvons remonter jusqu'à l'ancêtre commun le plus probable en cartographiant les caractéristiques connexes ; dans ce cas, la queue (tail), les oreilles (ears) et les moustaches (whiskers). C'est ce que nous appelons un arbre phylogénétique. Pour les virus, nous procédons un peu différemment. Nous séquençons leur génome, la cartographie lettre par lettre de tout ce qu'ils font. Nous examinons ensuite la similitude de deux génomes en les plaçant l'un à côté de l'autre et en comparant chaque lettre à chaque endroit pour voir si c'est la même. Ce processus s'appelle l'alignement des séquences.
Quelle est l’origine du SARS-CoV-2 ?
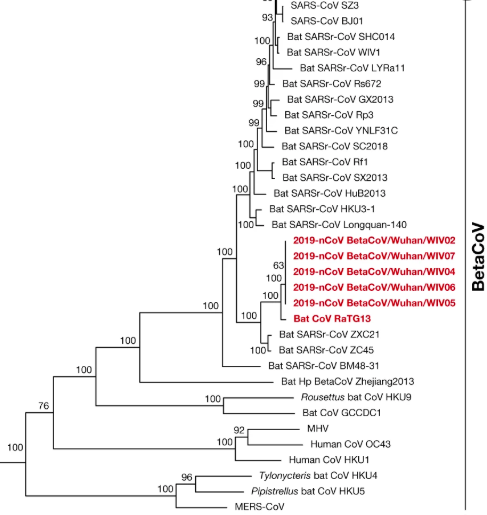
Une fois le nCoV-2019 (désormais appelé SARS-CoV-2) isolé des patients et entièrement séquencé, les scientifiques ont aligné son génome sur celui d'autres espèces de coronavirus précédemment identifiées. Ils ont découvert que le virus le plus proche du SRAS-CoV-2 était un coronavirus provenant de chauves-souris. Plus précisément, le Bat CoV RaTG13 est un coronavirus que l'on trouve chez l’espèce de chauve-souris Rhinolophus affinis.
Maintenant, nous pouvons examiner où, dans le génome, les virus sont le plus et le moins similaires. Commençons donc par examiner une carte du génome du SARS-CoV-2.

Une représentation visuelle du génome du SARS-CoV-2, avec l'aimable autorisation de Biorender. Nous pouvons voir la structure 3D de la protéine de spike et nous mettons en évidence une région particulière appelée domaine de liaison du récepteur (RBD). Il est montré lié à la protéine de surface humaine, ACE2.
Gardez la carte ci-dessus à l'esprit lorsque vous regardez ce qui suit :

En regardant le graphique ci-dessus, la ligne bleue (Bat CoV RaTG13) ressort. Il s'agit d'un coronavirus de chauve-souris, et vous pouvez voir qu'il a constamment une identité nucléotidique de 90 à 100 % (sur l'axe des y). Cela signifie que lorsque vous alignez ce Bat CoV RaTG13 avec le nouveau SARS-CoV-2 et que vous regardez le long du génome en comparant la séquence, ils sont très similaires à la plupart des endroits. En comparaison, le SARS-CoV original (CoV-SARS BJ01 en rouge) présente une similarité de 50 à 80 % par rapport au SARS-CoV-2 ; bien qu'il soit encore assez proche, il n'est pas aussi proche que le Bat CoV RaTG13.
Mais si vous regardez à nouveau attentivement la carte de similarité des nucléotides, vous remarquerez peut-être quelque chose de drôle. Il y a une forte baisse de la similarité de tous les virus de la région qui codent pour la protéine spike (autour de la position du nucléotide 23 000). Ce signal est le moins prononcé pour Bat CoV RaTG13, mais c'est quand même la chute la plus forte par rapport à d'autres parties du génome.
La protéine spike à la surface des coronavirus est la raison pour laquelle ces virus ont cette apparence. Voici une image des virions du SARS-CoV-2. Ces petits pics qui dépassent du virion forment une "couronne" semblable à celle du soleil et sont composés de la protéine spike. Cette protéine permet au virus de pénétrer dans les cellules humaines. Elle agit comme une clé qui ne fonctionne que sur des serrures spécifiques. La serrure est une protéine que l'on trouve à la surface de nombreuses cellules de notre corps, y compris celles des poumons. Cette protéine est appelée ACE2.
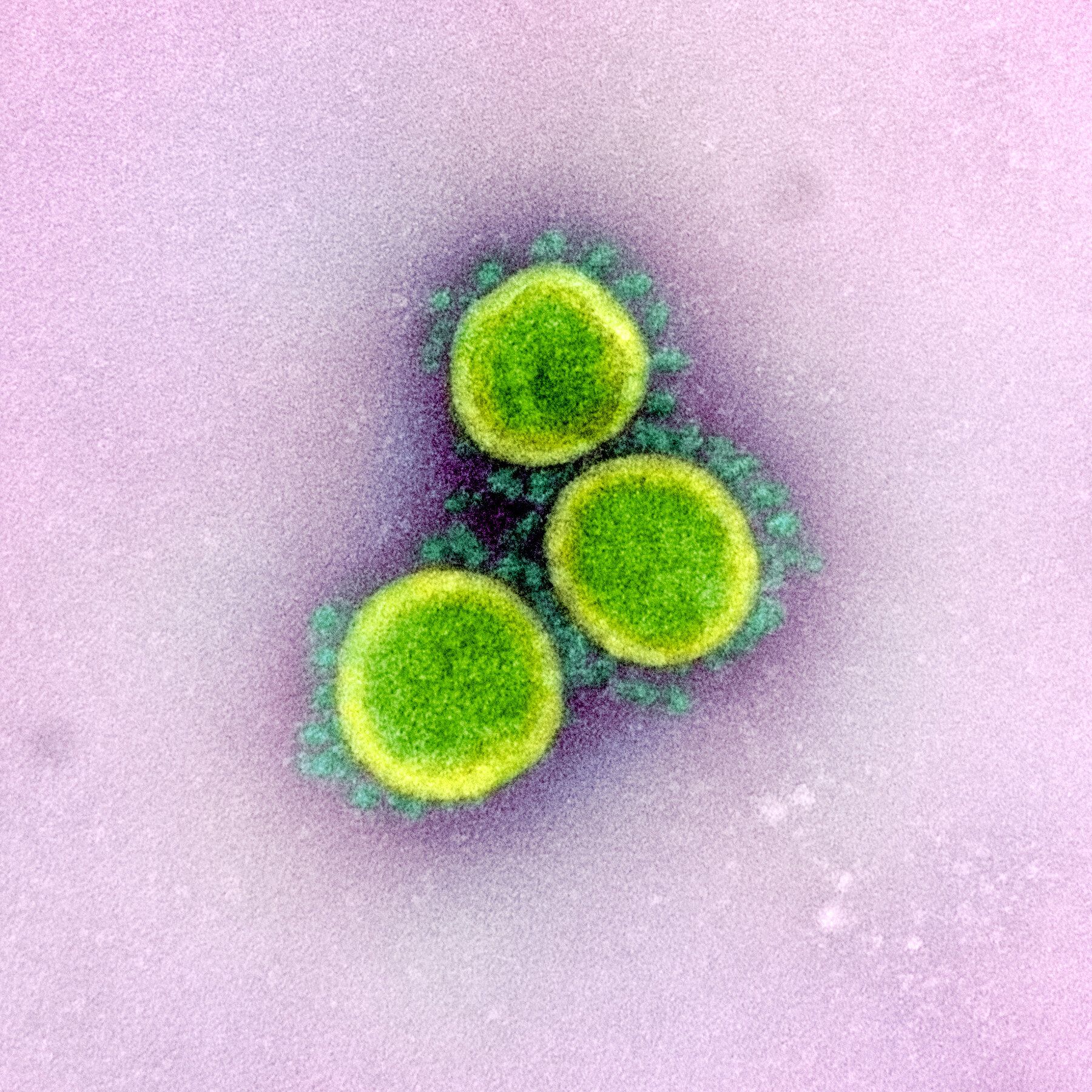
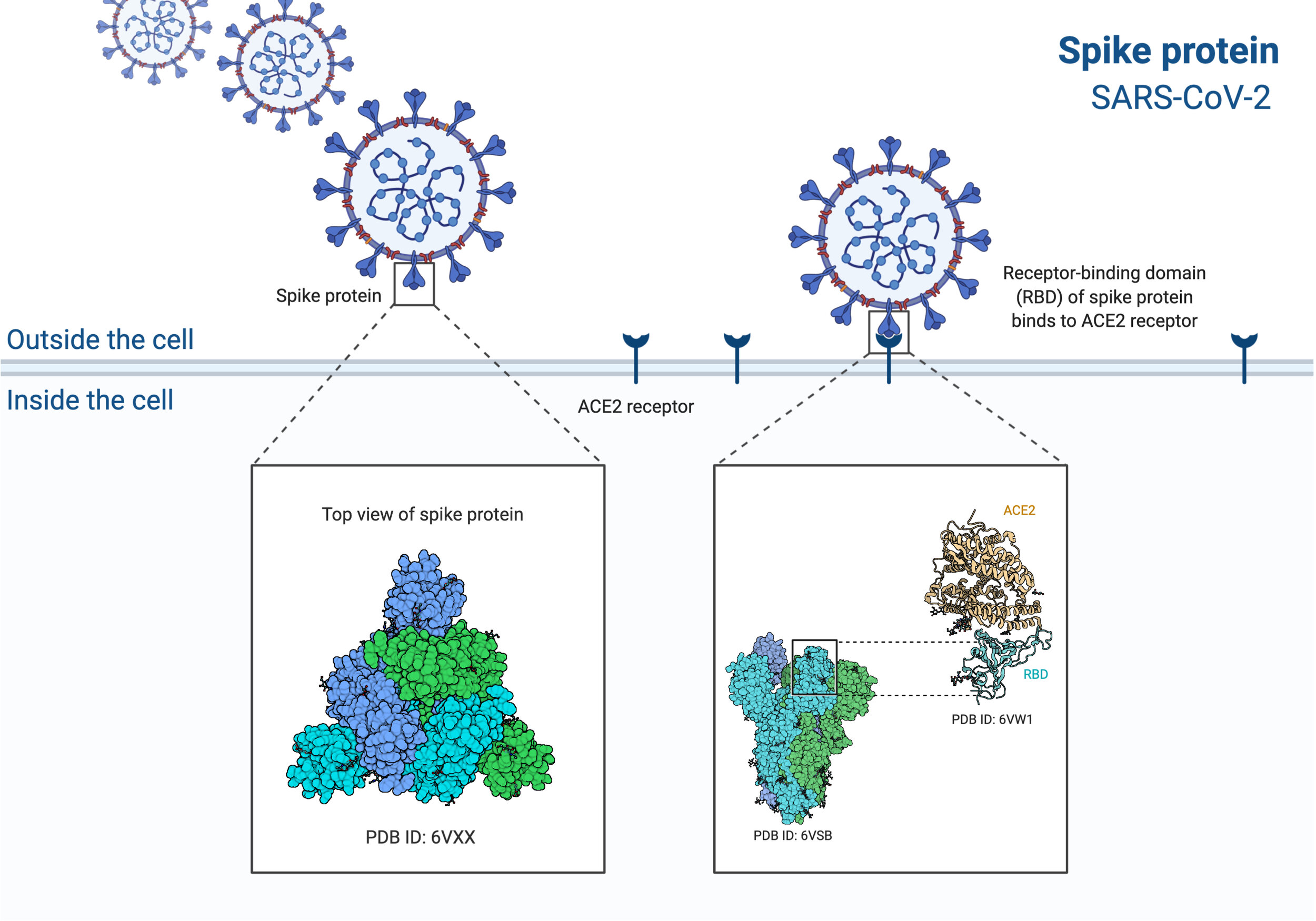
Image 1 : Micrographie à transmission électronique de particules du virus SARS-CoV-2, isolées d'un patient. Image capturée et colorée au centre de recherche intégré (IRF) du NIAID à Fort Detrick, Maryland. Crédit : NIAID
Image 2 : Les particules du virus SARS-CoV-2, se liant au verrou et à la clé de l'ACE2 à la surface de la cellule. Modifié à partir de BioRender.
Étant donné l'importance de la protéine spike pour les coronavirus, les différences de séquence entre RaTG13 et SARS-CoV-2 dans la région du de la protéine spike du génome sont critiques. Heureusement, un autre laboratoire a trouvé le seul virus ayant une séquence similaire à celle du SARS-CoV-2 dans le domaine de liaison au récepteur (RBD) du spike. Il est intéressant de noter que ce virus provenait de pangolins malais.

Firdia Lisnawati: AP Photo; image d’un pangolin malais
Ci-dessous, se trouve un autre graphique de similarité des nucléotides. Mais cette fois, le haut du graphique n'indique pas de similarité avec le SARS-CoV-2, mais plutôt une similarité avec un coronavirus trouvé dans les pangolins malaisiens. Les deux virus sur lesquels il faut se concentrer sont le SARS-CoV-2 en rouge et le Bat CoV RaTG13 en vert.
Comme on pouvait s'y attendre, les lignes rouges et le vertes se suivent, jusqu'à ce que soudainement, elles divergent. C'est dans la région orange qu'ils divergent et c'est là que le SARS-CoV-2 est le plus similaire au coronavirus que l'on trouve dans un pangolin malais. Il est important de noter que la région orange est la RBD (appelée ici le motif de liaison du récepteur) cruciale dans la protéine spike.

Figure modifiée d’après Xiao et al. 2020 https://doi.org/10.1101/2020.02.17.951335
Que signifie tout cela ? Nous avons maintenant une hypothèse pour l'espèce intermédiaire entre l'homme et la chauve-souris ... le pangolin !
Si vous vous souvenez, pour le SARS-CoV, l'espèce intermédiaire était la civette masquée. Pour le SARS-CoV-2, ce pourrait être le “ fourlimier écailleux”, un mammifère avec des écailles kératineuses, alias le Pangolin malais ou Pangolin javanais. Il est important de noter que le coronavirus du pangolin n’a pas une grande similarité sur l'ensemble du génome du SARS-CoV-2, mais seulement sur une région de la protéine spike appelée RBD (Receptor Binding Domain). Cela suggère qu'une recombinaison virale a pu avoir lieu, où un morceau du coronavirus pangolin a été transféré dans un coronavirus de chauve-souris, ce qui a donné un tout nouveau virus.
Qu'est-ce que la recombinaison et comment se produit-elle?
Les coronavirus peuvent subir un processus appelé "recombinaison homologue" (Lai et al. 1990 et Lai et al. 1997). Imaginons que deux souches de virus différentes, mais apparentées, entrent dans la même cellule au même moment. Appelons-les A et B. Il est possible que lorsque A commence à faire des copies de lui-même, dans une petite partie de son génome, il puisse accidentellement copier dans la région homologue, ou similaire, du virus B ! C'est comme si une personne utilisait une photocopieuse et qu'une deuxième personne échangeait un de ses feuilles de papier en plein milieu de la pile et que le tout était agrafé. Dans notre cas, le virus qui sort est presque identique au virus A original, mais il a une petite section qui semble identique au virus B. C'est peut-être ce qui s'est passé pour le SARS-CoV-2. La plus grande partie ressemble à un coronavirus de chauve-souris, à l'exception de la petite région RBD de la protéine spike où elle ressemble au coronavirus pangolin.
Notre hypothèse jusqu’à présent :
En utilisant des alignements de séquences qui comparent de nombreux génomes de virus les uns par rapport aux autres, nous avons une idée de l'origine du SARS-CoV-2. Il est probable que la plupart du SARS-CoV-2 proviennent d'un coronavirus de chauve-souris apparenté au RaTG13, mais qu'il a capté une partie de sa protéine spike lors d'une interaction avec un autre coronavirus chez un hôte intermédiaire. Notre principal suspect actuel ? Un pangolin. Il est possible que le nouveau coronavirus se soit passé par les chauves-souris et les pangolins avant de rejoindre les humains. Cependant, nous n'avons pas encore trouvé de preuve irréfutable. Les scientifiques doivent encore isoler un virus intact dans les pangolins qui contient tous ces morceaux ensemble - nous n'avons que des indices de la séquence SARS-CoV-2. Il est possible que notre hypothèse actuelle soit erronée. Cependant, au fur-et-à mesure que d'autres virus seront séquencés et que des données seront recueillies, nous pourrons reconstituer une histoire plus précise de l'émergence du SARS-CoV-2.
Nous espérons que vous avez apprécié ce premier article sur les origines potentielles du SARS-CoV-2. Dans notre prochain article, nous verrons comment des composants spécifiques du SARS-CoV-2 rendent ce virus si efficace pour infecter les humains. Restez à l'écoute et lavez-vous les mains !

Traduit en Français par Sébastien J. Puechmaille à partir de l’original de @csstevens91
Sébastien Puechmaille est Enseignant-Chercheur à l’Université de Montpellier où ses recherches se concentrent sur l’étude des chauves-souris et de leurs pathogènes (y compris les coronavirus).
http://batlab.ucd.ie/~spuechmaille/
Twitter: @Bats_Chiroptera & @SJPuechmaille

Christian Stevens
Christian, qui a obtenu sa licence au Harvey Mudd College (USA), est un étudiant en doctorat à l'école de médecine de Mount Sinai (USA).
Il a rejoint le laboratoire de Lee Benhur en 2018 et a depuis travaillé sur deux projets principaux. Le premier fait appel au génie sur des virus pour explorer l'utilisation du virus Sendai comme vecteur viral pour fournir des outils d'édition de gènes. Le second a impliqué un travail informatique de construction de pipelines pour l'analyse en utilisant à la fois le séquençage Illumina et le séquençage direct de l'ARN d'Oxford Nanopore. Les principaux intérêts de Christian ont été d'orienter la recherche clinique de classe mondiale vers les patients les plus marginalisés, en particulier dans les domaines des maladies infectieuses et de la virologie.
christian.stevens@icahn.mssm.edu
Twitter: @csstevens91