Los Orígenes del SARS-CoV-2: Primera Parte
El comienzo de una nueva especie

Imagen cortesía de MammalWatching.com
Naturalmente, una de las primeras preguntas que nos hacemos cuando aparece una nueva enfermedad es "¿de dónde vino esto?" En este artículo hablaremos sobre los orígenes del SARS-CoV-2 y su probable llegada a la población humana.
Con cada nuevo patógeno emergente hay una teoría de la conspiración sobre cómo se originó. SARS-CoV-2 no es la excepción. En la Primera Parte de esta breve serie, hablaremos sobre la evidencia de cómo surgió este virus en la población humana. En la Segunda Parte, profundizaremos un poco más y discutiremos porqué este virus es tan efectivo para infectar a los humanos. Y finalmente, en la Tercera Parte, tendremos todas las piezas necesarias para discutir y desacreditar el conjunto de teorías de conspiración que rodean al SARS-CoV-2.
La primera pregunta que debemos intentar responder es, ¿cuál es el origen más probable de SARS-CoV-2? Para responderla correctamente, necesitamos entender algunos antecedentes.
Animales y Virus que Mutan

Gráfico que describe la transmisión de enfermedades zoonóticas y cómo diferentes vectores pueden propagar la enfermedad (cortesía de la OMS).
Se estima que el 75 por ciento de los patógenos humanos emergentes son de origen zoonótico. "Zoonótico" se refiere a enfermedades que pasan de animales a humanos (ver imagen de arriba). Cada animal tiene sus propios microorganismos (virus, bacterias, etc.) que se han vuelto expertos en infectar a ese animal en particular. Por ejemplo, el Virus de la Inmunodeficiencia de Simios (SIVcpz por sus siglas en inglés) infecta de manera enérgica a los chimpancés. Sin embargo, es muy malo para infectar ratones. SIVcpz también lucha para infectar a los humanos, pero los científicos creen que SIVcpz fue el precursor del Virus de Inmunodeficiencia Humana (VIH-1). A medida que SIVcpz saltó entre especies, comenzó a adquirir mutaciones que permitieron que el virus proliferara en humanos, hasta que se convirtió en una especie distinta de virus: el VIH. Este proceso de transmisión y mutación zoonótica es importante para entender los orígenes del SARS-CoV-2.
Los microorganismos están hechos para los animales que infectan. Sin embargo, es importante recordar que en la naturaleza, estos microorganismos no se están diseñando con previsión [al animal que van a infectar]. Al contrario, buscan al azar mientras producen billones de copias de sí mismos, cada una ligeramente diferente a la anterior. La mayoría de estos cambios no ayudan en lo absoluto, y algunos incluso pueden ser dañinos para el microorganismo, pero con el tiempo tropiezan con algo realmente ingenioso que les permite ser más eficientes. Este proceso se llama selección natural. Los virus se replican (es decir, hacen más virus a partir de sí mismos) de manera rápida y descuidada, cometiendo errores, pero también mejorando las posibilidades de que adquieran una mutación ventajosa que les permita superar a todos los demás.
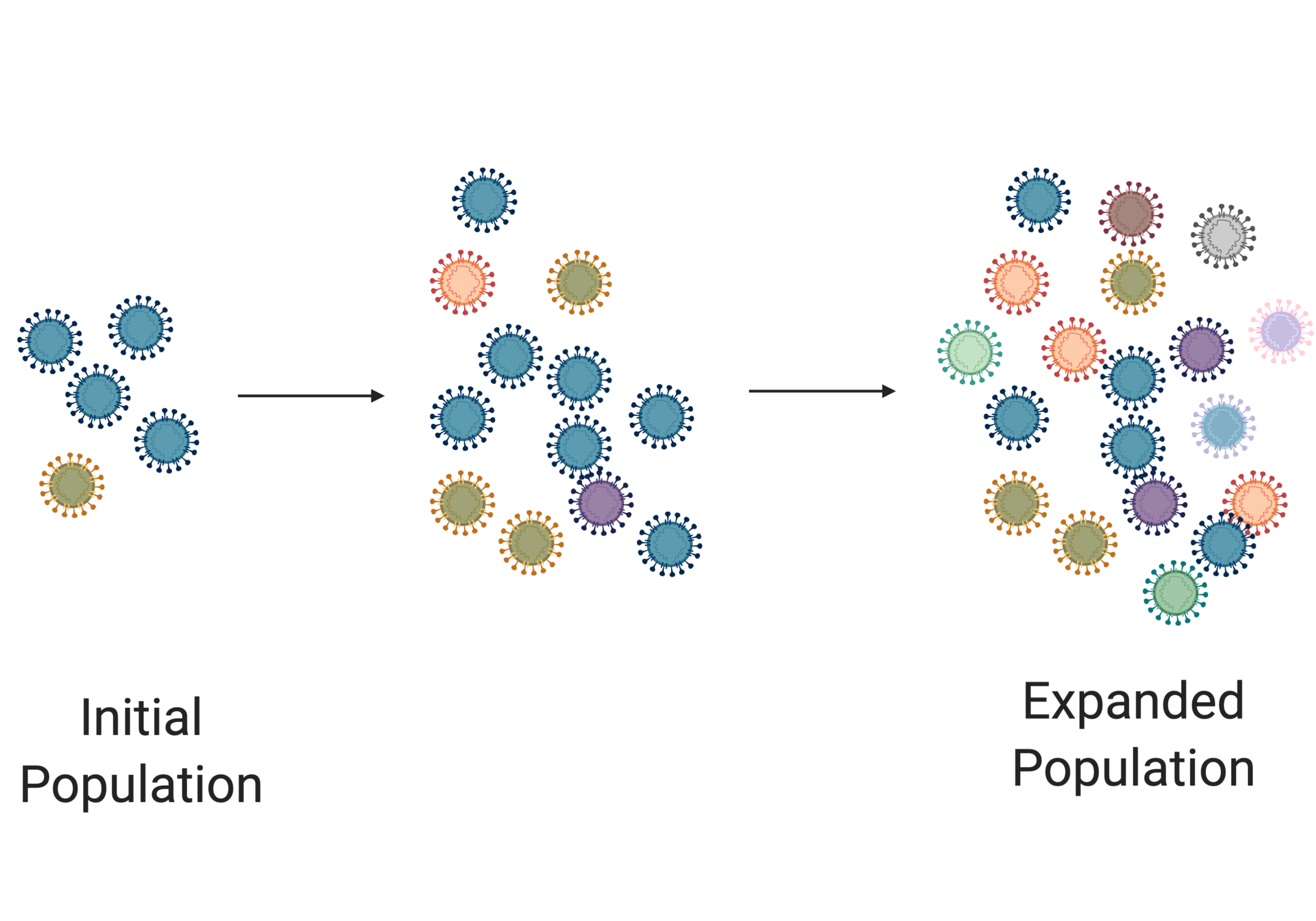
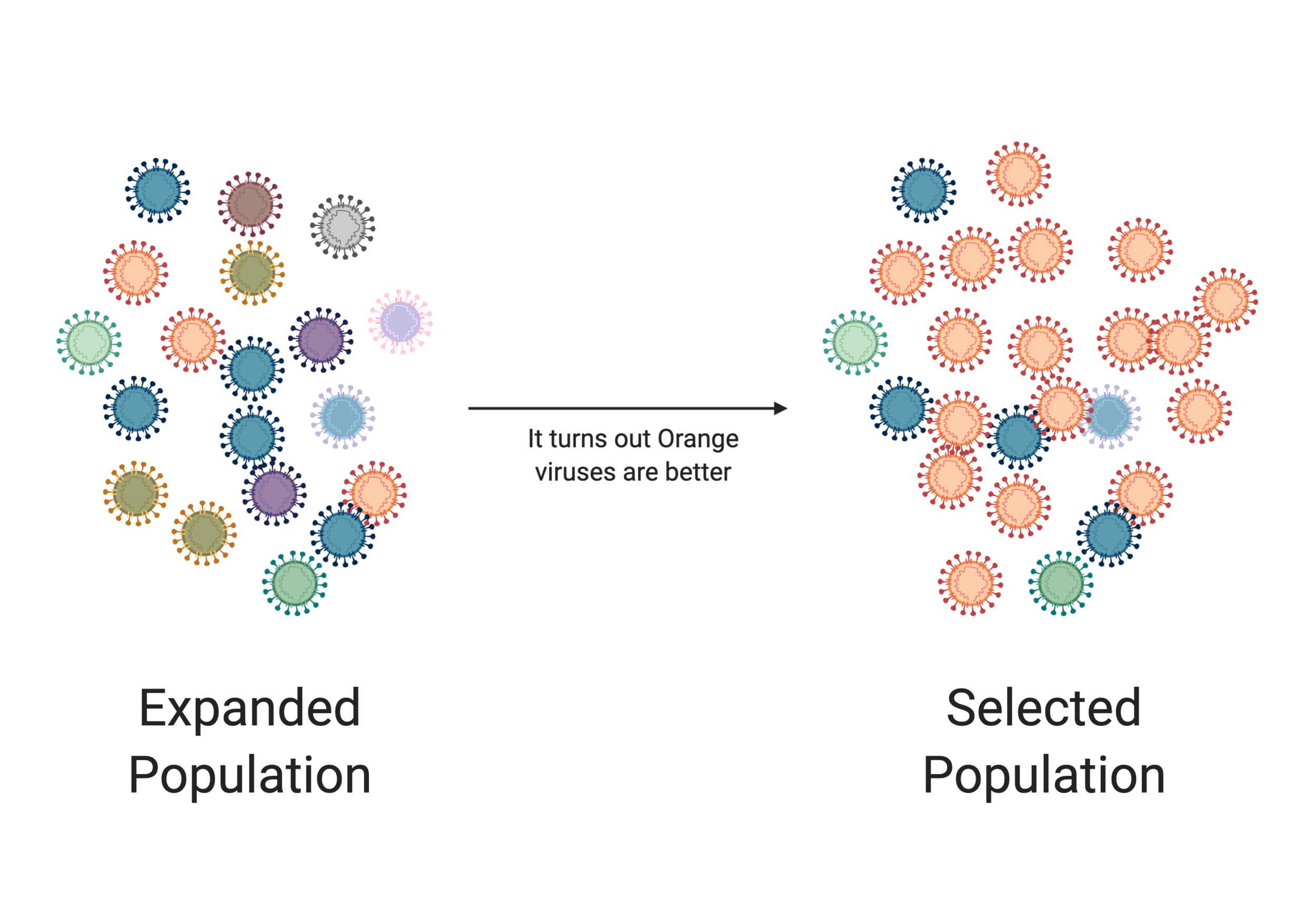
En la imagen de arriba vemos una población inicial de virus. Son pocos y no muy diversos (como indican los diferentes colores). Pero a medida que estos virus se replican, comienzan a cometer errores. A medida que la población se expande, vemos un aumento en la diversidad de la población de virus. Pero en la siguiente imagen, ocurre algo que le da una ventaja al virus naranja (por ejemplo, saltó a una nueva especie). En este escenario, el virus naranja domina en la población. Este proceso de expansión y selección es vital para la evolución viral.
La mayoría hemos aprendido anteriormente sobre la selección natural, pero es importante subrayar este punto. Este proceso de mutación aleatoria que resulta en virus que son cada vez más adecuados es en lo que pensamos cuando tratamos de averiguar de dónde proviene un virus. Podemos evaluar el plan para SARS-CoV-2, su genoma de ARN, y buscar pistas sobre su origen. Un virus tendrá escrita dentro de su historia genética la firma de cualquier sitio donde haya estado.
Primeros Indicios
Antes de que se reconocieran los primeros casos de SARS-CoV-2 había cuatro especies de virus del género Betacoronavirus (el mismo al que se uniría el SARS-CoV-2) que se sabía infectaban a los humanos. Los dos primeros, HKU1 y OC43, causan un resfriado común. Los otros dos, SARS-CoV y MERS-CoV, son virus extremadamente letales que han causado brotes graves (pero no pandemias globales).


Izquierda: titulares de 2003, durante el primer brote de SRAS. Derecha: titular de 2012, cuando surgió MERS. Los contagios de MERS desde camellos y camélidos hacia humanos continúan hasta nuestros días.
El 31 de diciembre del 2019 el gobierno chino informó que había un grupo de casos de neumonía viral en la ciudad de Wuhan, China. Muchos de los casos estaban relacionados con el mercado mayorista de mariscos de Huanan (Huanan Seafood Wholesale Market). El primer temor era que el SARS-CoV original había regresado, o que tal vez estaba circulando una nueva cepa mortal de influenza. En una semana los científicos chinos ya habían presentado al mundo toda la secuencia genómica del virus para que el mundo la viera. Aprendimos que esto no era SARS-CoV, sino un nuevo coronavirus inicialmente llamado nCoV-2019.
En ese momento, el mundo sabía tres cosas sobre el virus:
Podía causar una neumonía viral potencialmente mortal.
Estaba estrechamente relacionado con el SARS-CoV.
Muchos de los casos originales, pero no todos, estaban vinculados al mercado mayorista de mariscos de Huanan.

Este es un gato civeta, el supuesto hospedero intermedio entre murciélagos y humanos en el brote original de SRAS de 2003. Cortesía de Kalyan Varma y Wikimedia Commons.
La investigación del brote original de SARS-CoV mostró que el virus probablemente se extendió a los humanos desde los murciélagos de herradura a través de un huésped intermedio, la civeta de palma enmascarada (foto a la derecha), antes de saltar a los humanos. Por lo tanto, los científicos plantearon la hipótesis de que nCoV-2019 (también conocido como SARS-CoV-2) también se propagó de los murciélagos a los humanos, tal vez utilizando un hospedero intermedio diferente en el proceso.
Aunque es una hipótesis prometedora, todavía necesitamos una forma de probarla. Eso nos lleva a la siguiente pregunta importante: ¿cómo determinamos los orígenes de un virus?
Las Herramientas de un Detective de Virus
Identificar los orígenes de un virus es un emocionante juego de deducción, acertijos lógicos e inferencias a lo Sherlock Holmes. Como detectives de virus, primero recopilamos la mayor cantidad posible de datos y evidencia. Desafortunadamente, a menudo no hay una evidencia irrefutable, por lo que debemos utilizar el razonamiento inductivo para llegar a conclusiones sobre los eventos que no hemos observado directamente.
Entonces, si no podemos ver un virus en el momento en que salta a un humano por primera vez, ¿cómo vemos de dónde vino un virus? Le damos el antiguo tratamiento 23andMe y observamos su genoma para comprender a sus antepasados.
Recuerde, como este virus hace copias de sí mismo, puede cometer errores. Algunos de estos errores dañarán al virus, y los virus que contienen esos errores fallarán. Otros errores ayudan al virus y esos virus crecerán abundantemente. Algunos de los errores no tendrán efecto sobre el virus. A estos últimos errores los llamamos mutaciones neutrales y, por consiguiente, todos los virus que son hijos de estos virus con errores contienen una mutación distintiva que indica su relación. Podemos usar estas mutaciones distintivas para construir un árbol genealógico.
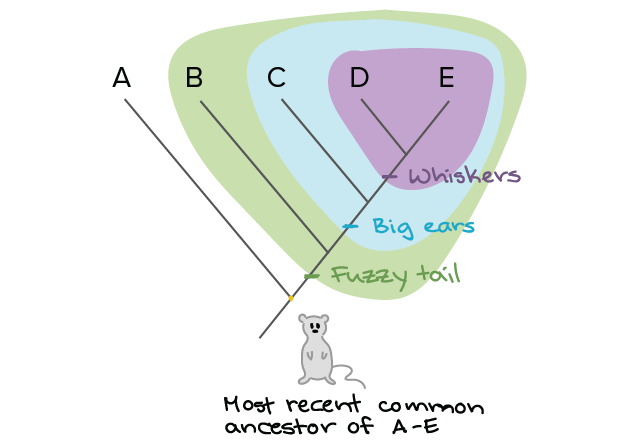
Una ilustración del árbol filogenético, cortesía de Khan Academy.
Como se ilustra arriba, al mapear características relacionadas (en este caso, al cola, las orejas y los bigotes) podemos ir hacia atrás hasta el ancestro común más probable. Llamamos a esto un árbol filogenético. Para los virus, esto lo hacemos un poco diferente. Secuenciamos su genoma, el plano letra por letra de todo lo que hacen. A continuación, vemos qué tan similares son dos genomas colocándolos uno al lado del otro y comparando cada letra en cada posición para ver si es la misma. Este proceso se llama alineación de secuencia.
¿Cuáles son los Orígenes del SARS-CoV-2?
Una vez que nCoV-2019 (ahora llamado SARS-CoV-2) se aisló de los pacientes y se secuenció por completo, los científicos alinearon su genoma con los genomas de otras especies de coronavirus previamente identificadas. Descubrieron que el pariente más cercano del SARS-CoV-2 era un coronavirus de los murciélagos; específicamente Bat CoV RaTG13, un coronavirus que se encuentra en la especie de murciélago herradura Rhinolophus affinis. Ahora podemos ver en qué parte del genoma los virus son más similares y menos similares. Así que primero veamos un mapa del genoma del SARS-CoV-2.

Una representación visual del genoma del SARS-CoV-2, cortesía de Biorender. Podemos ver la estructura 3D de la proteína espiga y destacamos una región particular llamada dominio de unión al receptor (RBD). Se muestra unido a la proteína de superficie humana, ACE2.
Tenga en cuenta el mapa anterior al mirar lo siguiente:

Mirando el gráfico anterior, se destaca la línea azul (Bat CoV RaTG13). Este es un coronavirus de murciélago, y puede ver en el eje y que tiene entre 90-100% de identidad de nucleótidos. Esto significa que cuando alineas este Bat CoV RaTG13 contra el nuevo SARS-CoV-2 y caminas a lo largo del genoma comparando las secuencias, ambas son muy similares en la mayoría de los sitios. En comparación, el SARS-CoV original (SARS-CoV BJ01 en rojo) varía entre una coincidencia del 50-80% en comparación con el SARS-CoV-2; aunque está bastante relacionado, no está tan cerca como Bat CoV RaTG13.
Pero si observa detenidamente el mapa de identidad de nucleótidos puede notar algo intrigante. Hay una fuerte disminución en la similitud de todos los virus en la región que codifica la proteína espiga (alrededor de la posición de nucleótido 23.000). Esta disminución es menos pronunciada para Bat CoV RaTG13, pero sigue siendo la caída más pronunciada en comparación con otras partes del genoma de este virus.
La proteína espiga en la superficie de los coronavirus es la razón por la cual estos virus se ven como se ven. A continuación se muestra una imagen de los viriones SARS-CoV-2. Esas pequeñas espigas que sobresalen del virión forman una "corona" similar a la del sol y están compuestas de proteína espiga. Esta proteína permite que el virus ingrese a las células humanas. Actúa como una llave que solo funciona en cerraduras específicas. La cerradura es una proteína que encontramos en la superficie de muchas células de nuestro cuerpo, incluidas las del pulmón. Esta proteína se llama ACE2.
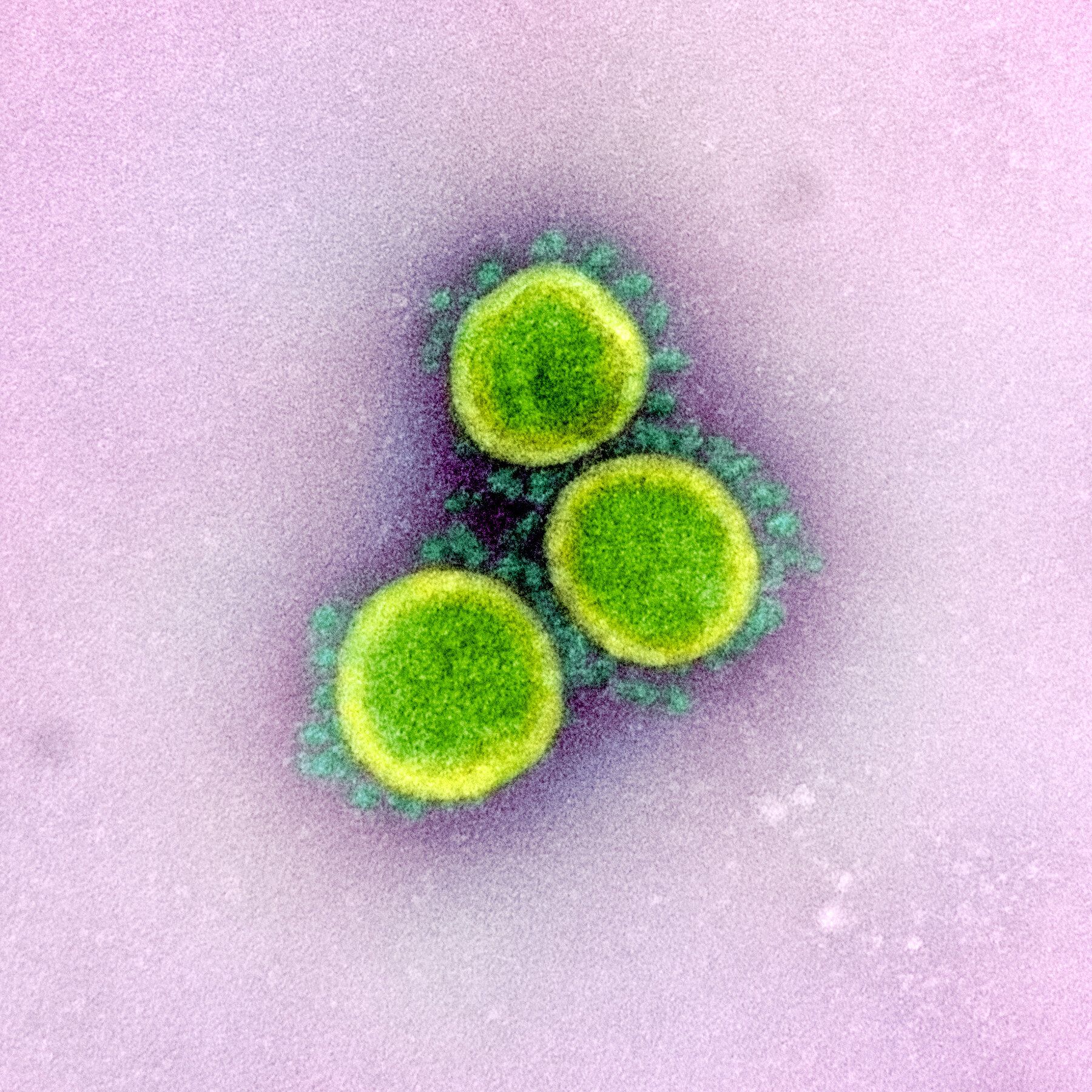
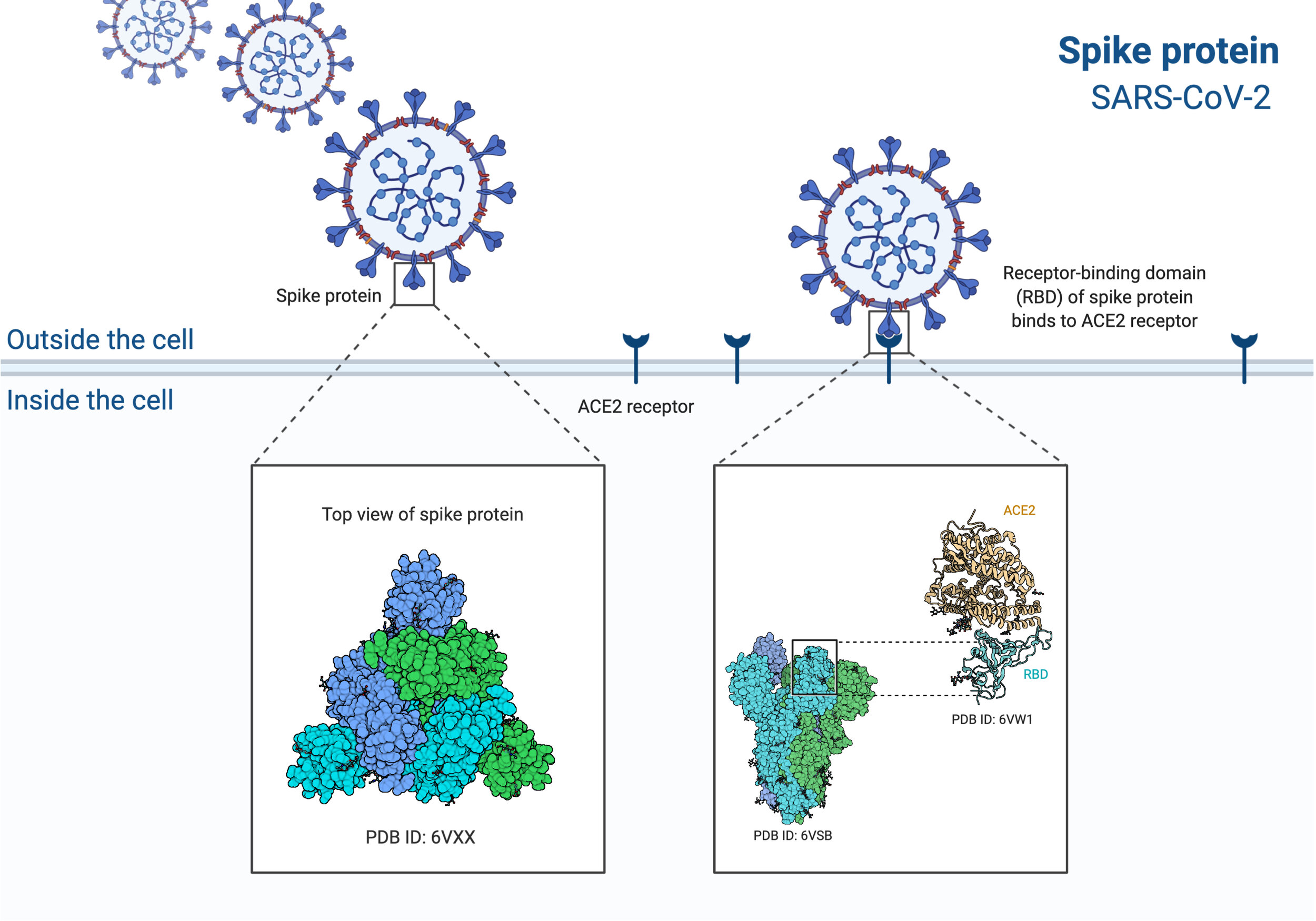
Imagen 1: Micrografía electrónica de transmisión de partículas del virus SARS-CoV-2, aisladas de un paciente. Imagen capturada y mejorada en color en el Centro de Investigación Integrada (IRF) del NIAID en Fort Detrick, Maryland. Crédito: NIAID
Imagen 2: Partículas del virus SARS-CoV-2, con la unión llave y cerradura para ACE2 en la superficie celular. Modificado de BioRender.
Dada la importancia de la proteína espiga para los coronavirus, las diferencias de secuencia entre RaTG13 y SARS-CoV-2 en la región espiga del genoma son cruciales. Afortunadamente, otro laboratorio encontró el único virus con una secuencia similar al SARS-CoV-2 en el dominio de unión al receptor (RBD) de la espiga. Curiosamente, este virus proviene de pangolines de Malasia.

Firdia Lisnawati: Foto AP; imagen de un pangolín malayo
Abajo hay otro gráfico de similitud de nucleótidos. Pero en esta ocasión la parte superior del gráfico no indica similitud con el SARS-CoV-2, sino similitud con un coronavirus encontrado en los pangolines de Malasia. Los dos virus en los que hay que enfocarse son SARS-CoV-2 en rojo y Bat CoV RaTG13 en verde.
Como era de esperar, vemos que el rojo y verde verde coinciden casi perfectamente hasta que, de repente, no lo hacen. Es en la región naranja donde difieren, y es aquí donde el SARS-CoV-2 es más similar al coronavirus encontrado en un pangolín malayo. Es importante destacar que la región naranja es el RBD crucial (denominado aquí motivo de unión al receptor) en la proteína de la espiga.

Figura alterada de Xiao et al. 2020 https://doi.org/10.1101/2020.02.17.951335.
¿Qué significa todo esto? Ahora tenemos una hipótesis para las especies intermedias entre humanos y murciélagos ... ¡el pangolín!
Si recuerdas, para el SARS-CoV la especie intermedia era la civeta de palma enmascarada. Para el SARS-CoV-2 puede ser el "oso hormiguero escamoso", un mamífero con escamas de queratina, también conocido como pangolín. Es importante destacar que el coronavirus pangolín no coincide con el genoma completo del SARS-CoV-2, sino que solo coincide con una región de la proteína espiga llamada RBD (dominio de unión al receptor). Esto sugiere que puede haber ocurrido una recombinación viral, donde una parte del coronavirus pangolín se transfirió a un coronavirus murciélago, creando un nuevo virus.
¿Qué es la recombinación y cómo ocurre?
Los coronavirus pueden someterse a un proceso llamado "recombinación homóloga" (Lai et al. 1990 y Lai et al. 1997). Imaginemos que dos cepas de virus diferentes, pero relacionadas, entraron en la misma célula en el mismo momento. Llamemos a los virus A y B. ¡Existe la posibilidad de que a medida que A comience a hacer copias de sí mismo, copie accidentalmente en una pequeña parte de su genoma la región homóloga o similar del genoma del virus B! Es como si una persona estuviera usando una fotocopiadora y una segunda persona intercambiara uno de sus hojas de papel justo en el medio de la pila y todo quedara engrapado. En nuestro caso, el virus que sale es casi idéntico al virus original A, pero tiene una pequeña sección que luce idéntica al virus B. Esto puede ser lo que sucedió con el SARS-CoV-2. La mayor parte se parece a un coronavirus de murciélago, excepto la pequeña región RBD de la proteína espiga, donde se parece al coronavirus pangolín.
Nuestra hipótesis hasta ahora:
Usando alineamientos de secuencia que comparan muchos genomas de virus entre sí tenemos una idea de dónde se originó el SARS-CoV-2. Es probable que la mayor parte del SARS-CoV-2 se derive de un coronavirus de murciélago relacionado con RaTG13, pero que tomó parte de su proteína espiga de una interacción con otro coronavirus en un huésped intermedio. ¿Nuestro principal sospechoso actual? Un pangolín. Una posibilidad es que el nuevo coronavirus se movió a través de murciélagos y pangolines antes de saltar a los humanos. Sin embargo, aún no hemos encontrado una evidencia irrefutable. Los científicos aún no han aislado un virus intacto en pangolines que contenga todas estas piezas juntas; solo tenemos indicios de la secuencia SARS-CoV-2. Es posible que nuestra hipótesis actual sea incorrecta. Sin embargo, a medida que se secuencian más virus y se recopilan datos, podemos reconstruir una historia más precisa de la aparición de SARS-CoV-2.
Esperamos que haya disfrutado este primer artículo sobre los posibles orígenes del SARS-CoV-2. En nuestra próxima publicación abordaremos cómo los componentes específicos del SARS-CoV-2 hacen que este virus sea tan bueno para infectar a los humanos. ¡Estén atentos y lávense las manos!
¡Esta traducción hecha los laboratorios Dávalos y Rojas!
Laboratorio de Rojas
Website: rojasdanny.wordpress.com
Twitter: @danny_cu
Laboratorio de Dávalos
online, Google scholar publications
twitter: @LabDavalos
skype: vampyrops
zoom: https://stonybrook.zoom.us/my/lmdavalos
office: 631.632.1554


Christian Stevens
Christian es un estudiante de doctorado en la Escuela de Medicina Mount Sinai y obtuvo su licenciatura en el Harvey Mudd College.
Se unió al Laboratorio de Benhur Lee en el 2018 y desde entonces ha trabajado en dos proyectos principales. El primero utiliza la ingeniería viral para explorar el uso del virus Sendai como un vector viral para entregar herramientas de edición de genes. El segundo ha involucrado el trabajo computacional en la construcción de procesos para el análisis utilizando tanto la secuenciación Illumina como la secuenciación directa de ARN de Oxford Nanopore. Los principales intereses de Christian han sido enfocar la investigación clínica de clase mundial hacia los pacientes más marginados, especialmente en los campos de enfermedades infecciosas y virología.
christian.stevens@icahn.mssm.edu
Twitter: @csstevens91