The Origins of SARS-CoV-2: Part 1

Image courtesy of MammalWatching.com
Naturally, one of the first questions we ask ourselves when a new disease appears is “where did this come from?” In this post, we’ll talk about the origins of SARS-CoV-2 and how it likely emerged in the human population.
With every new emerging pathogen, there is a conspiracy origin theory to go with it. SARS-CoV-2 is no different. In Part 1 of this short series we’ll talk about the evidence for how this virus emerged into the human population. In Part 2, we’ll dig a bit deeper and discuss why this virus is so effective at infecting humans. And finally, in Part 3, we’ll have all the pieces we need to discuss and debunk the bevy of conspiracy theories surrounding SARS-CoV-2.
The first question we must attempt to answer is, what is the most likely origin of SARS-CoV-2? To answer this correctly, we’ll need a bit of background.
Animals and Mutating Viruses

Graphic describing the transmission of zoonotic diseases and how different vectors can spread the disease (courtesy of the WHO).
It’s estimated that 75 percent of emerging human pathogens are zoonotic in origin. ‘Zoonotic’ refers to diseases that pass from animals to humans (see above image). Every animal has its own microorganisms—viruses, bacteria, etc.—that have become proficient at infecting that particular animal. For example, Simian Immunodeficiency Virus (SIVcpz) robustly infects chimpanzees. However, it is very bad at infecting mice. SIVcpz also struggles to infect humans, but scientists believe that SIVcpz was the precursor to Human Immunodeficiency Virus (HIV-1). As SIVcpz jumped species, it started to acquire mutations that allowed the virus to grow well in humans instead, until it became a separate species of virus—HIV. This process of zoonotic transmission and mutation is important for understanding the origins of SARS-CoV-2.
Microorganisms are built for the animals they infect. However, it’s important to remember that, in nature, these microorganisms aren’t designing themselves with forethought. Instead, they grasp around at random, making trillions of copies of themselves with slight differences each time. The majority of these don’t help at all and some may even hurt, but eventually they stumble on something really nifty, and it allows them to be more efficient. This process is called natural selection. Viruses replicate (ie make more of themselves) in a fast and sloppy manner, making mistakes but also improving the chances they acquire an advantageous mutation that outcompetes all others.
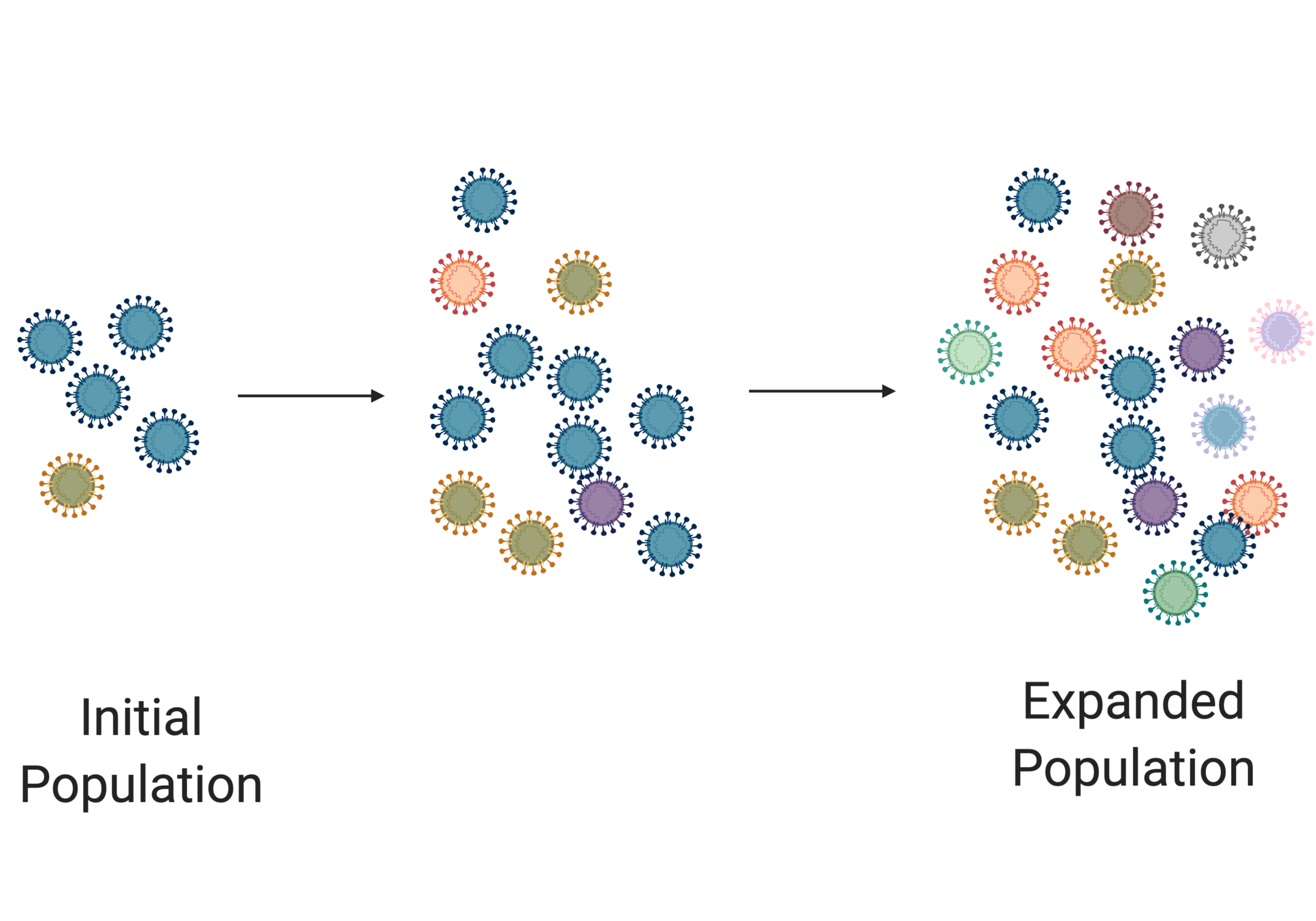
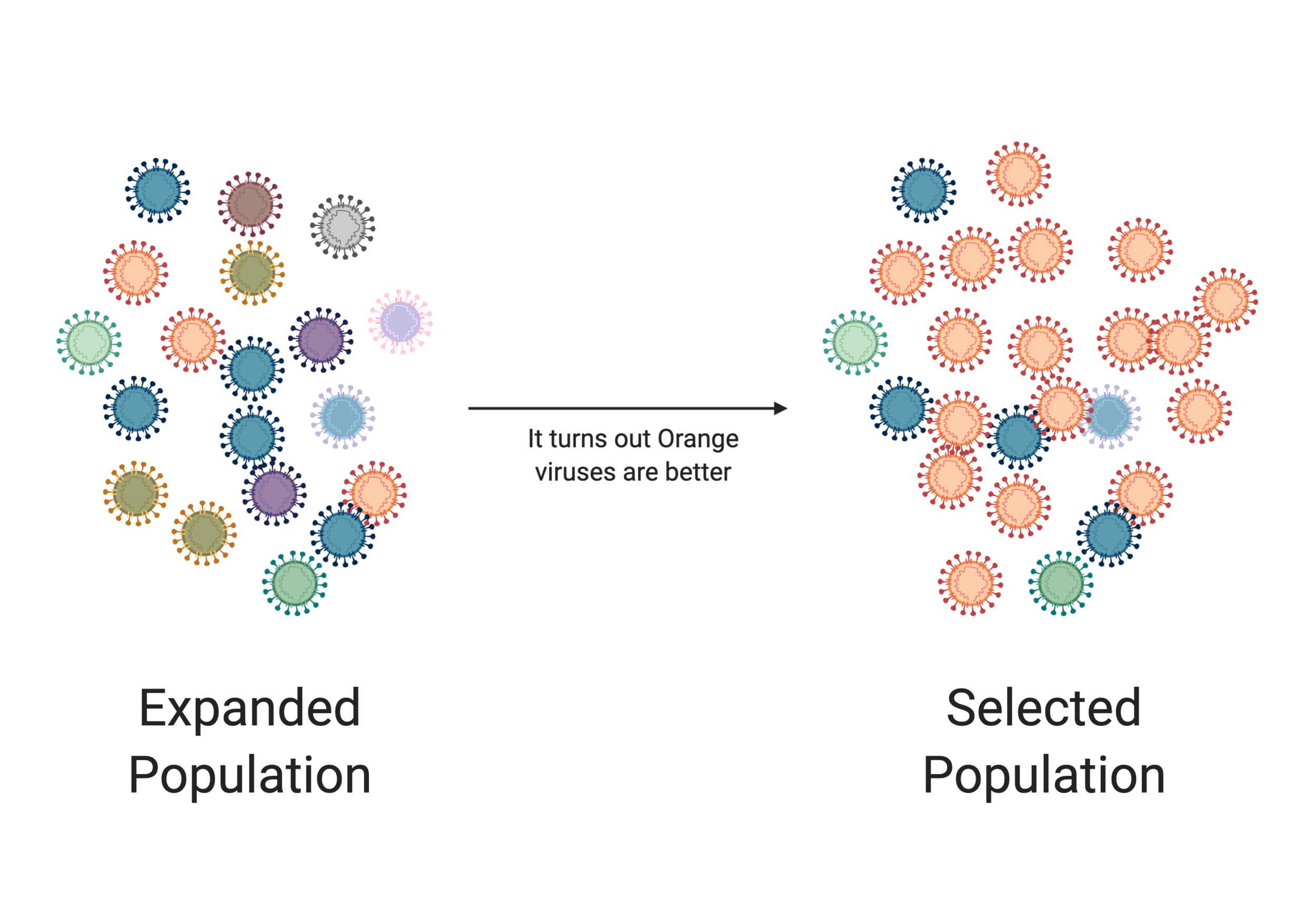
In the image above we see an initial population of viruses. They are small in number and not very diverse (denoted by their different colors). But as these viruses replicate, they start making mistakes. As the population expands, we see an increase in the diversity of the virus population. But in the next image, something occurs that gives an advantage to the orange virus (eg it jumped into a new species). In this scenario, the orange virus takes over the population. This process of expansion and selection is vital for viral evolution.
Most of us have learned about natural selection before, but it’s important to stress the point. This process of random mutation resulting in increasingly fit viruses is what we think about when we try to figure out where a virus comes from. We can assess the blueprint for SARS-CoV-2, its RNA genome, and look for hints as to its origin. Anywhere that a virus has been will have its signature written within its genetic history.
The First Hints
Before the first cases of SARS-CoV-2 were recognized, there were 4 species of viruses known to infect humans in the genus Betacoronavirus (the same that SARS-CoV-2 would join). The first two, HKU1 and OC43, cause a common cold. The other two—SARS-CoV and MERS-CoV— are extremely lethal viruses that have caused serious outbreaks (but not global pandemics).


Left - headlines from 2003, during the first SARS outbreak. Right - headline from 2012, when MERS first emerged. MERS spillovers from camels and camelids into humans continue to this day.
On December 31, 2019, the Chinese government reported a cluster of cases of viral pneumonia in the city of Wuhan, China. Many of the cases were connected to the Huanan Seafood Wholesale Market. The first fear was that the original SARS-CoV was back again, or perhaps there was a new, deadly strain of influenza circulating. Within a week, Chinese scientists had already submitted the entire genomic sequence of the virus for the world to see. We learned that this was not SARS-CoV, but a novel coronavirus initially named nCoV-2019.
At this moment, the world knew 3 things about the virus:
It could cause a potentially deadly viral pneumonia
It was closely related to SARS-CoV
Many, but not all of the original cases were linked to the Huanan Seafood Wholesale Market

This is a civet cat, the presumed intermediate host between bats and humans for the original 2003 SARS outbreak. Courtesy of Kalyan Varma and Wikimedia Commons.
Research from the original SARS-CoV outbreak showed the virus likely spilled over into the humans from horseshoe bats via an intermediate host; the masked palm civet (pictured right); before jumping into humans. Thus, scientists hypothesized that nCoV-2019 (aka SARS-CoV-2) also spilled over from bats to humans, perhaps using a different intermediate host in the process.
Although a promising hypothesis, we still need a way to test it. That brings us to the next important question: how do we determine the origins of a virus?
The Tools of a Virus Detective
Identifying the origins of a virus is an exciting game of Holmesian deduction, logic puzzles, and inferences. As virus detectives, we first gather as much data and evidence as possible. Unfortunately, there is often no smoking gun, so we must use inductive reasoning to arrive at conclusions about events we have not observed directly.
So if we can’t watch a virus in the moment it jumps to a human for the first time, how do we see where a virus came from? We give it the old 23andMe treatment and look at its genome to understand its ancestors.
Remember, as this virus makes copies of itself, it can make mistakes. Some of these mistakes will hurt the virus and viruses containing those mistakes will fail. Some of these mistakes help the virus and those viruses will grow abundantly. Some of these mistakes will have no effect on the virus. We call these neutral mutations and as a result, all viruses that are children of these now contain a signature mutation indicating their relatedness. We can use these signatures to build a family tree.
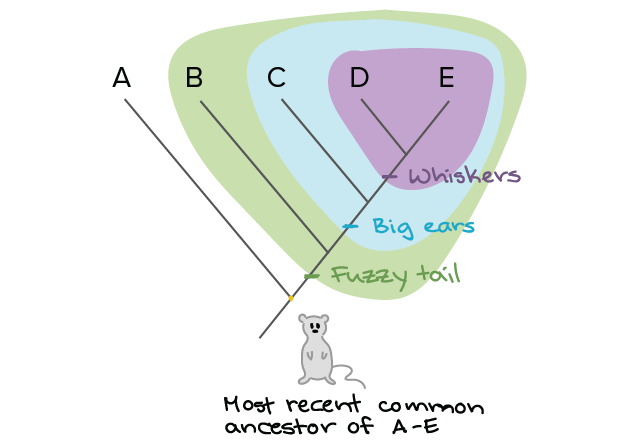
A phylogenetic tree illustration, courtesy of Khan Academy
As illustrated above, we can work backwards to the most likely common ancestor by mapping related features; in this case the tail, ears, and whiskers. We call this a phylogenetic tree. For viruses we do this a little differently. We sequence their genome, the letter-by-letter blueprint of everything they do. We next look at how similar two genomes are by setting them next to each other and comparing every letter at every spot to see if it’s the same. This process is called sequence alignment.
What are the Origins of SARS-CoV-2?
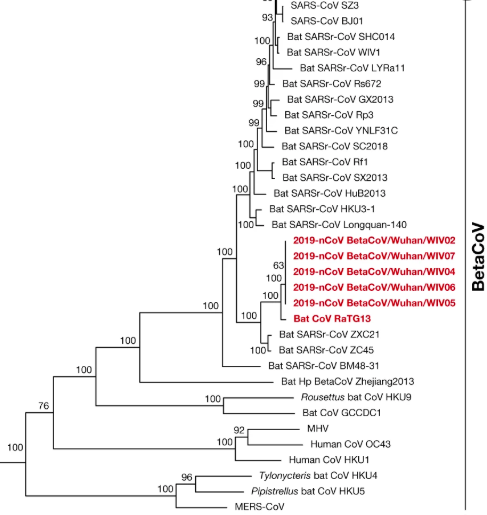
Once nCoV-2019 (now called SARS-CoV-2) was isolated from patients and fully sequenced, scientists aligned its genome to other previously identified species of coronaviruses. They discovered the closest relative of SARS-CoV-2 was a coronavirus from bats. Specifically, Bat CoV RaTG13 is a coronavirus found in the horseshoe bat species Rhinolophus affinis.
Now, we can look at where in the genome the viruses are most and least similar. So let’s first look at a map of the SARS-CoV-2 genome.

A visual representation of the SARS-CoV-2 genome, courtesy of Biorender. We can see the 3D structure of the spike protein and highlighted is a particular region called the receptor binding domain (RBD). It is shown bound to the human surface protein, ACE2.
Keep the above map in mind when looking at the following:

Looking at the above graph, the blue line (Bat CoV RaTG13) stands out. This is a bat coronavirus, and you can see that it tracks along at between 90-100% nucleotide identity (on the y-axis). This means that when you line up this Bat CoV RaTG13 against the new SARS-CoV-2 and walk along the genome comparing the sequence, they are very similar at most spots. In comparison, the original SARS-CoV (SARS-CoV BJ01 in red) hovers between a 50-80% match compared to SARS-CoV-2; while it is still quite related, it is not as close as Bat CoV RaTG13.
But if you look carefully again at the nucleotide identity map, you may notice something funny. There is a sharp drop-off in the similarity of all viruses in the region that codes for the spike protein (around nucleotide position 23,000). It’s the least pronounced for Bat CoV RaTG13, but it’s still the steepest drop compared to elsewhere in the genome.
The spike protein on the surface of coronaviruses is why these viruses look the way they do. Below is an image of SARS-CoV-2 virions. Those little spikes protruding from the virion form a Sun-like “corona” and are composed of spike protein. This protein allows the virus to enter human cells. It acts like a key that only works on specific locks. The lock is a protein we find on the surface of many cells in our body, including those in the lung. This protein is called ACE2.
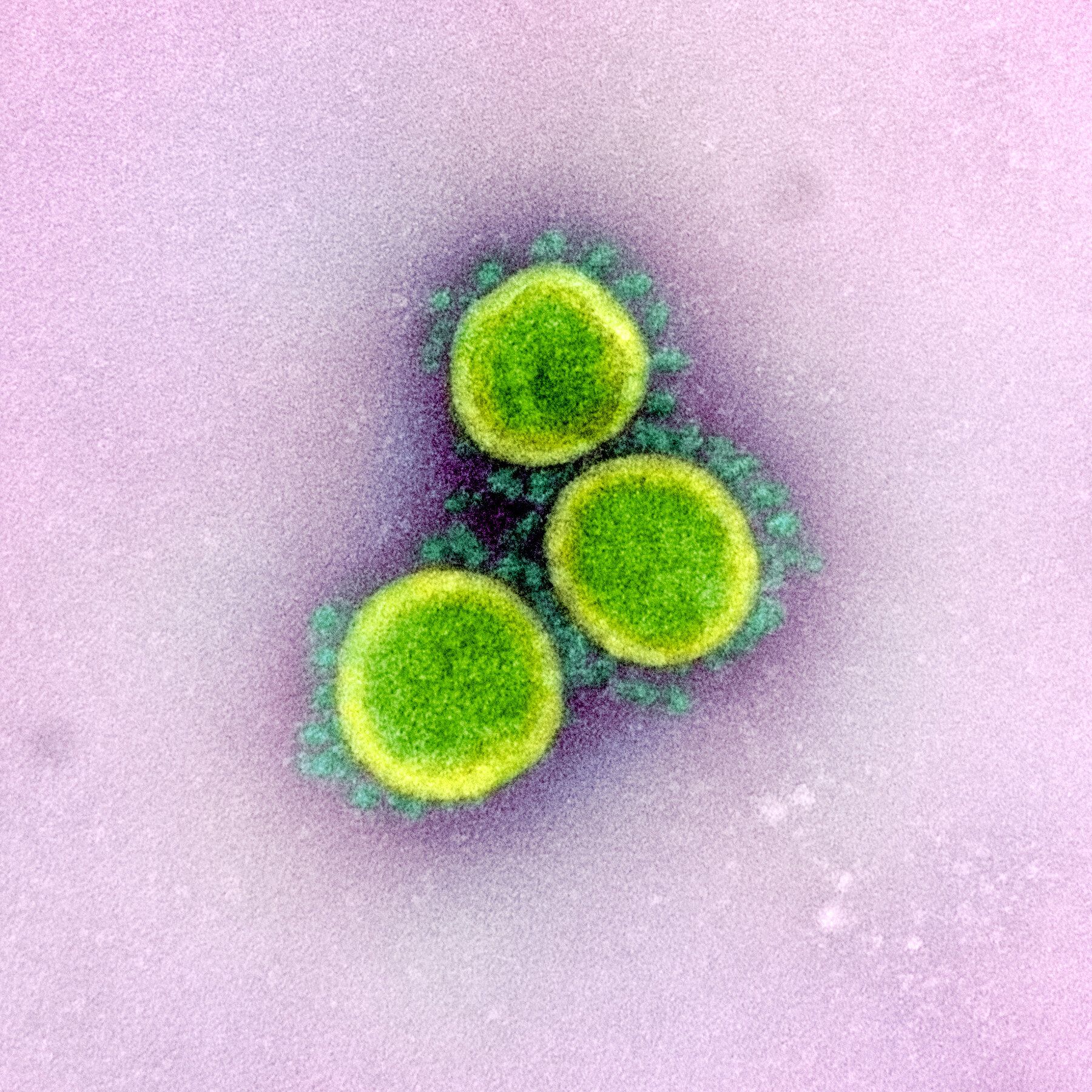
Image 1: Transmission electron micrograph of SARS-CoV-2 virus particles, isolated from a patient. Image captured and color-enhanced at the NIAID Integrated Research Facility (IRF) in Fort Detrick, Maryland. Credit: NIAID
Image 2: The SARS-CoV-2 virus particles, binding lock and key to ACE2 on the cell surface. Modified from BioRender.
Given the importance of the spike protein for coronaviruses, the sequence differences between RaTG13 and SARS-CoV-2 in the spike region of the genome are critical. Fortunately, another lab found the only virus with a similar sequence to SARS-CoV-2 in the receptor binding domain (RBD) of spike. Interestingly, this virus came from Malayan pangolins.

Firdia Lisnawati: AP Photo; image of a Malayan pangolin
Below, is another nucleotide similarity graph. But this time, the top of the graph doesn't indicate similarity to SARS-CoV-2, but instead similarity to a coronavirus found in Malayan pangolins. The two viruses to focus on are SARS-CoV-2 in red and Bat CoV RaTG13 in green.
As expected we see red and green almost perfectly matched up until suddenly, they're not. It is in the orange region where they diverge and it's here that SARS-CoV-2 is most similar to the coronavirus found in a malayan pangolin. Importantly, the orange region is the crucial RBD (referred to here as the receptor binding motif) in the spike protein.

Figure altered from Xiao et al. 2020 https://doi.org/10.1101/2020.02.17.951335.
What does all of this mean? We now have a hypothesis for the intermediate species between humans and bats…the pangolin!
If you remember, for SARS-CoV the intermediate species was the masked palm civet. For SARS-CoV-2 it may be the “scaly anteater”, a mammal with keratin scales, aka the pangolin. Importantly, the pangolin coronavirus does not match the entire genome of SARS-CoV-2, but only matches a region of the spike protein called the RBD (Receptor Binding Domain). This suggests that viral recombination may have occurred, where a piece of the pangolin coronavirus was transferred into a bat coronavirus, making a brand new virus.
What is recombination and how does it occur?
Coronaviruses can undergo a process called “homologous recombination” (Lai et al 1990 and Lai et al. 1997). Let’s imagine two different, but related virus strains happened to enter the same cell at the same moment. Let’s call them A and B. There is a chance that as A begins to make copies of itself, in one small part of its genome it may accidentally copy in the homologous, or similar, region from the virus B! It’s like one person is using a copy machine and a second person swapped in one of their pieces of paper right in the middle of the stack and it all got stapled together. In our case, the virus that comes out is near identical to the original virus A, but has one small section that looks identical to virus B. This may be what happened for SARS-CoV-2. Most of it looks like a bat coronavirus except the small RBD region of the spike protein where it resembles pangolin coronavirus.
Our hypothesis so far:
Using sequence alignments that compare many virus genomes against each other, we have an idea of where SARS-CoV-2 originated. It’s likely that most of SARS-CoV-2 is derived from a bat coronavirus related to RaTG13, but that it picked up part of its spike protein from an interaction with another coronavirus in an intermediate host. Our current prime suspect? A pangolin. One possibility is that the new coronavirus moved through bats and pangolins before jumping to humans. However, we have not yet found a smoking gun. Scientists have yet to isolate an intact virus in pangolins that contains all of these pieces together—we only have hints from the SARS-CoV-2 sequence. It is possible our current hypothesis is incorrect. However, as more viruses are sequenced and data are collected, we can piece together a more accurate story of SARS-CoV-2 emergence.
We hope you enjoyed this first post discussing the potential origins of SARS-CoV-2. In our next post, we address how specific components in SARS-CoV-2 make this virus so good at infecting humans. And in Part 3 we deal directly with the some of the alternative origin theories. Stay tuned and wash those hands!

Christian Stevens
Christian is an MD/PhD Student at the Mount Sinai School of Medicine who got his BS from Harvey Mudd College.
He joined the Benhur Lee Lab in 2018 and has since worked on two main projects. The first uses viral engineering to explore the use of Sendai Virus as a viral vector to deliver gene editing tools. The second has involved computational work building pipelines for analysis using both Illumina sequencing and Oxford Nanopore direct-RNA sequencing. Christian’s main interests have been in directing world class clinical research towards the most marginalized patients, especially in the fields of infectious disease and virology.
christian.stevens@icahn.mssm.edu
Twitter: @csstevens91