The Origins of SARS-CoV-2: Part 2

Case courtesy of Dr Fabio Macori, Radiopaedia.org. From the case rID: 74867
Part 1 of this miniseries focused on where SARS-CoV-2 originated. Part 2 addresses what makes it different, and why it’s so good at infecting people. Armed with this knowledge, we will be able to address the fear: is this from a lab?
A Review of Part 1
If you haven’t read it yet, I recommend checking out Part 1. We wrote about how scientists investigate the origins of a virus. From there, we discussed our leading hypothesis for the origins of SARS-CoV-2. It has two parts:
Most of the viral genome likely comes from a coronavirus closely related to a horseshoe bat coronavirus called Bat CoV RaTG13.
The all-important receptor binding domain (RBD) of the Spike protein is closely related to an entirely different coronavirus found in Malayan pangolins.
We also addressed an important result from this likely recombination event: the new RBD for SARS-CoV-2 is ten times better than SARS-CoV. As a reminder, the RBD, in part, dictates how well the spike protein gets SARS-CoV-2 into our cells by binding a human protein on cell surfaces called ACE2. The new RBD shows a 10x increase in its binding affinity for ACE2.
So before starting Part 2, we already have one piece of the puzzle explaining why SARS-CoV-2 is so effective at infecting humans—it is very good at latching on to the human protein (ACE2) needed to invade our cells.
How SARS-CoV-2 Enters Cells: Activating the Spike Protein
In the YouTube video above, you can see the spike protein from SARS-CoV-2. It starts with a top down view of the protein, then spins around the side, ending back at the top. Something important about this protein is that it functions in a trimer. This means that three identical copies of the protein (light grey, dark grey, and blue, in the video above) come together to perform the protein’s function. As you watch it rotate, you might notice that something looks slightly off about the blue monomer. It has a little piece that is flipped up at the top of the protein. In fact, this is the RBD getting ready to bind to human ACE2.
Before we go further, here’s some quick biochemistry background. Proteins aren’t static—that is, they’re not stuck in the same shape once they are made. Different things can cause them to modify their structure. In the case of SARS-CoV-2 spike, a piece of the protein must be cut to activate it. Think of it like pressing the button to release your umbrella. By pressing the button you move a latch and pop! The umbrella takes its full “active” shape. Cutting the spike protein allows it to pop open into its infectious shape. This activation is called proteolytic cleavage (literally, “protein-breaking cutting”) and it is necessary before SARS-CoV-2 can bind ACE2 for entry. Many other viruses—like HIV, influenza, and Dengue viruses— must also cut their envelope/spike proteins to be activated for viral entry.
One more biochemistry fact: proteins that cut (cleave) other proteins are called proteases. To make sure that proteases don’t just start cutting blindly, they only recognize specific cleavage sites in the sequence of their target proteins. This sequence tells the protease which protein to cut, where to cut it, and even sometimes when to do so. But why is this important?
A Spike Protein’s Secret Weapon:
The Polybasic Cleavage Site
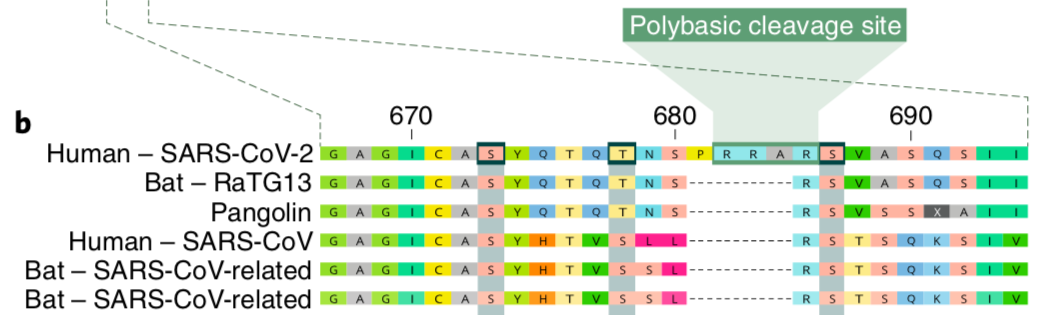
The amino acid sequence for the cleavage sites of the most closely related viruses to SARS-CoV-2. Edited from Andersen et al 2020.
In the picture above, the amino acid (protein) sequences of a small region of the spike protein from six different coronaviruses are lined up. You might recognize the closest relative of SARS-CoV-2, the Bat CoV RaTG13, as well as the sequence for the pangolin coronavirus. This image also includes the original SARS-CoV and two additional bat coronaviruses. This is all done to illustrate a simple point; SARS-CoV-2 is the only sequence with a polybasic cleavage site. We can see that SARS-CoV-2 is unique without even knowing what a polybasic cleavage site is. Where every other virus has a gap (dashed lines), SARS-CoV-2 spike has an extra chunk of amino acids (labelled P R R A). These four extra amino acids turn out to be quite important for SARS-CoV-2’s success.
The polybasic cleavage site
Polybasic cleavage site is a mouthful. We already discussed that a cleavage site tells proteases where to cut our proteins (and in the case of Spike, this means it is activated). The original SARS-CoV, and other coronaviruses, have a monobasic cleavage site that lacks those extra “R” amino acids in the P R R A. Simply put, a polybasic cleavage site is an easier target for proteases to find. Proteins with a polybasic cleavage site can be cut by the protease furin which is found in many tissue types. On the flip-side is a monobasic cleavage site. Proteases that target this site may be limited to a much smaller subset of tissues and locations in the body. Altogether this means:
If the Spike protein contains a polybasic cleavage site, then the virus may be able to grow and wreak havoc in more tissue types.
For example, in avian influenza (among many other viruses) where highly pathogenic strains have a polybasic cleavage site, viruses can grow in many tissues aside from the birds’ lungs and gut, causing an increase in disease severity (virulence).

Here we see “R” which indicates the monobasic cleavage site and “R X R/K R” which indicates the polybasic cleavage site. Blue stars indicate what tissues can be infected (modified from Han et al. 2020).
So the takeaway? The polybasic cleavage (ie furin activation) site in the spike protein may increase the transmission and/or disease severity of SARS-CoV-2.
Can SARS-CoV-2 Hide?
We’re still learning about how the human immune system handles SARS-CoV-2 infection. One way our immune system can do this is to find a target for antibodies on SARS-CoV-2. But what if the virus could find a way to evade detection? Right now, there is some evidence that infected humans can produce virus-neutralizing antibodies, and a suggestion that such an antibody response might prevent re-infection (at least, in macaques). However, we don’t know how good these antibody responses are overall. Let’s take a moment to refresh ourselves on how antibodies can work to block infection.

Antibodies blocking the binding of Spike to ACE2, preventing cell entry. Created by Christian Stevens, courtesy of Biorender.
When an antibody binds its protein target, it’s incredibly effective at blocking that target. But, antibodies can’t fight what they can’t bind. Successful viruses have different strategies for dealing with this - including “hiding” their target proteins from antibodies (HIV) or even changing what the targets look like over time (influenza). SARS-CoV-2 doesn’t appear to be changing very fast, so what might help “hide” it from antibodies?
O-Linked Glycan
An o-linked glycan is a sugar molecule that is attached to the surface of a protein. In our bodies, these are sometimes used like a flag on the protein to indicate something such as blood type. Other times they help a protein maintain its structure, like in antibodies. Curiously, right next to the polybasic cleavage site on the SARS-CoV-2 spike, there is also a new o-linked glycan site.
One hypothesis is that the glycans create a sort of shield that “hides” the spike protein from being recognized by antibodies. When our immune system searches for important sites for antibodies to target, viruses that avoid those antibodies have a selective advantage. Viruses don’t do this consciously (remember our natural selection explainer in Part 1?). As antibodies start blocking all the viruses where they can see the target, any virus that makes a mutation resulting in an o-linked glycan site near the target area shields it from detection by our immune system. This is known as a glycan shield.

Modified from Karsten and Alter 2017. The yellow clusters indicate glycans that hide the green protein backbone from antibodies.
So what makes SARS-CoV-2 so good at infecting and spreading among humans?
Its spike protein is very good at binding human ACE2 to enter our cells (see Part I)
The spike protein can be activated in a wide range of cells and tissues (polybasic cleavage site)
The spike protein might be able to hide from antibodies (glycan shield)
How and When Did SARS-CoV-2 Develop These Tools?
Most of the SARS-CoV-2 genome is similar to other closely-related viruses found in bats or pangolins, and so we understand the origins of how spike binds ACE2 so well. However, the polybasic cleavage site and the O-linked glycan addition site are tools only SARS-CoV-2 has. At what stage could the virus have acquired these features?
Hypothesis 1: Before spillover into humans
Hypothesis 2: While amplifying (ie replicating) in humans
Spoiler: We haven’t found the smoking gun sequence to answer this question yet.
If hypothesis 1 is correct, then somewhere in the wild, there is a reservoir of viruses that look very similar to SARS-CoV-2. Whether it is in pangolins, bats, or some other intermediate animal, there may be other SARS-like viruses that already have the tools to spillover into humans again. If we knew this to be true, we could take action now to develop surveillance programs and create potential drugs and vaccines in preparation for such an event.
If hypothesis 2 is correct, then when SARS-CoV-2 first interacted with humans, it probably replicated and spread less well (like MERS does). But over some replication cycles where the virus made mistakes, some mutants gained the polybasic cleavage site and the glycan shielding. In doing so, they acquired the advantage in the natural selection game and took off to spread among humans. If this was the case, then we can implement better measures to monitor humans for new infections, and catch such spillover viruses before they “get good” at spreading among humans.
We are still in the very early days of SARS-CoV-2 basic science research, and it will take the sequencing of other coronaviruses in the wild before we discover the precise origin story of these particular mutations. But discovering the answer to this question will help guide our plans for preventing or managing the next Coronavirus outbreak before it reaches the severity we see with SARS-CoV-2 today.
For most scientists, the story stops here until we’ve done more research. We’ve got some leads on animal reservoirs, we’ve got information on mechanisms of transmission and pathogenesis in humans, and we have some ideas on how to answer the next scientific questions. Most scientists will look at this so far and say we’re on the right track because:
All the data gathered so far fits our hypotheses
Our hypotheses are the most parsimonious and best satisfy Occam’s Razor
Our hypotheses are testable and falsifiable
This strongly implies that the virus was not designed or modified in a lab
However, the internet is full of folks who aren’t scientists, and whose questions don’t quite fit the scientific mold. And it’s important to consider alternative theories and not simply brush anything away. In Part 3, we’ll directly examine some of the other theories and fears surrounding the origins of this new coronavirus; ‘til then, we hope that Parts 1 and 2 have already helped arm you with scientifically rigorous answers!
Check our Part 3 where we start calling out alternative theories directly now that we have the tools to engage them.

Christian Stevens
Christian is an MD/PhD Student at the Mount Sinai School of Medicine who got his BS from Harvey Mudd College.
He joined the Benhur Lee Lab in 2018 and has since worked on two main projects. The first uses viral engineering to explore the use of Sendai Virus as a viral vector to deliver gene editing tools. The second has involved computational work building pipelines for analysis using both Illumina sequencing and Oxford Nanopore direct-RNA sequencing. Christian’s main interests have been in directing world class clinical research towards the most marginalized patients, especially in the fields of infectious disease and virology.
christian.stevens@icahn.mssm.edu
Twitter: @csstevens91